
川村所長の勉強会参加記録
2019.07.05
血糖/体重コントロールをめざす2型糖尿病の薬物療法
2019年6月24日
演題「血糖と体重コントロールの両立をめざす2型糖尿病の薬物療法」
演者: 東京医科歯科大学 山田 哲也先生
場所:ホテル、ニューグランド
内容及び補足「
糖尿病の治療の留意点としては以下のものが挙げられる。
1. できるだけ良い血糖トントロール
2. 重症低血糖が無いこと
3. 体重増加が無いこと
である。
エネルギー摂取量と消費量の差が脂肪細胞へ蓄積され体重増加となる。
消費エネルギーの内訳を見てみると、基礎代謝に60%、反応性熱産生で10%、身体活動が30%と考えられている。
褐色脂肪組織(Brown adipose tissue:BAT)
古典的には、齧(ゲッ)歯類、特に冬眠動物で発達しており、肩甲骨間や腋窩、腎周囲に褐色脂肪細胞塊として存在しており、褐色脂肪細胞の分化と組織形成は胎仔期に完成する。
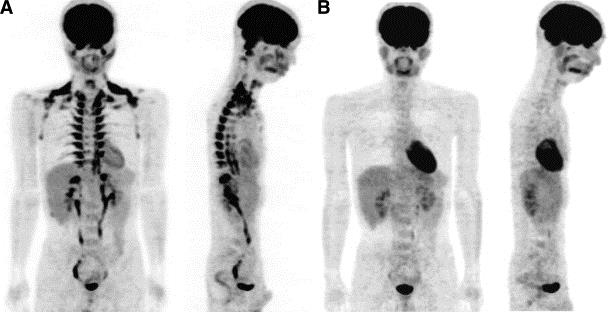
陽電子画像診断法(fluorodeoxyglucose-positron emission tomography:FDG-PET)を用いて、24℃の環境にいた後と、27℃の環境にいた後での撮像の違いを見ると、FDGの脂肪組織への取り込みの違いが解る。
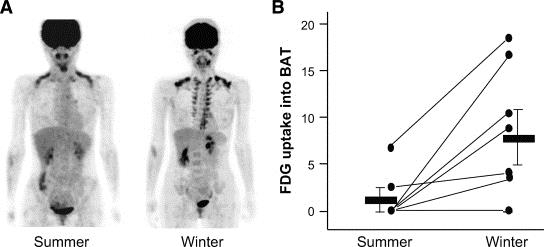
BATへのFDGの取り込みは夏と冬でも異なり、冬において、肩甲骨間、腎周囲、鎖骨上窩、腋窩部、傍椎体部にあるBATの熱産生機能が亢進していることがわかる。
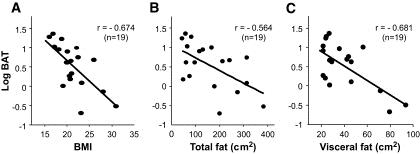
BATにおける寒冷により活性化するFDGの取り込み量はBMIや内臓脂肪量と逆相関する。
組織学的にUCP1陽性に染まる脂肪細胞が鎖骨城下の脂肪織から得られる。
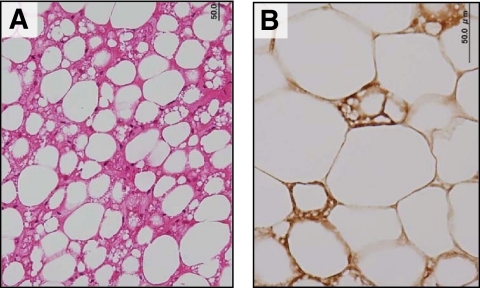
Histological identification of UCP1-positive brown adipocytes in fat depots obtained from the supraclavicular region. Tissue sections were stained with hematoxylin and eosin (A) or anti-serum against rat UCP1 (B).
Diabetes. 2009 Jul; 58(7): 1526-1531.
参:ベージュ脂肪細胞:ベージュ脂肪細胞は形態学的に似ているが細胞内に多房性脂肪滴を持ち、特異的蛋白質UCP1を発現したミトコンドリアに飛んでいる。単房性脂肪滴を持ち細胞質に乏しい白色脂肪細胞とは対照的である。機能的には、余剰エネルギーを中性脂肪として貯蔵する白色脂肪細胞とは異なり、褐色脂肪細胞とベージュ脂肪細胞はUCP1が酸化的リン酸化を脱共役させることにより熱産生を行う。マウスでは褐色脂肪細胞は肩甲骨間や腎周囲に細胞塊を形成して局在するのに対して、ベージュ脂肪細胞は鼠径部などの白色脂肪細胞組織中に誘導的かつ散財的に存在する。
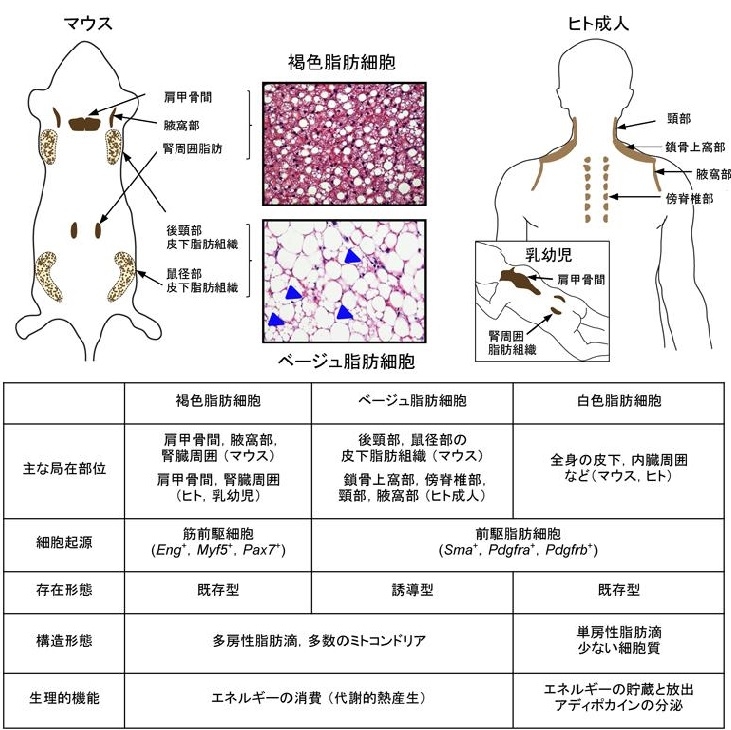
この現象は、白色脂肪の褐色化(Browning of white fat)と呼ばれる。発生起源として、褐色脂肪細胞は骨格筋と共通するMyobenic gactor 5(Myf5)を発現する筋前駆細胞に由来するのに対し、ベージュ脂肪細胞は白色脂肪細胞同様Myf5陰性で、platelet-derived growth factor receptor a (PDGFRa)やSmooth muscle actin(SMA)を発現する前駆脂肪細胞に由来する。
近年、陽電子画像診断法(fluorodeoxyglucose-positron emission tomography:FDG-PET)を用いた研究により、人でも一定量BATが肩甲骨間、腎周囲、鎖骨上窩、腋窩部、傍椎体部などに存在することが明らかになった。(Diabetes. 2009 Jul; 58(7): 1526-1531.)
このうち肩甲骨間のBATの遺伝子発現パターンはマウスの古典的褐色脂肪細胞とよく似ていることから、ヒト肩甲骨間のBATは古典的褐色脂肪細胞により構成されていると考えられている。この肩甲骨間BATは新生児や乳幼児で認められるが、成人では認められない。
成人のBATは鎖骨上窩部や腋窩部、傍脊椎部に分布している。鎖骨上窩部のBATから単一クローンに由来する細胞株の遺伝子検討では、遺伝子発現プロファイルがマウスのベージュ細胞と酷似しているので、成人が持つBATは主にベージュ細胞により構成されていると考えられる。(Cell Metab. 2015;22 997-1008)
成人のBAT活性が夏に最小になり、冬に最大となる誘導性及び可塑性を持つ。なお、人では、褐色脂肪細胞腫で構成される組織と、ベージュ脂肪細胞で構成されている組織の両者が総じてBAT(褐色脂肪組織)と呼ばれている。
https://seikagaku.jbsoc.or.jp/10.14952/SEIKAGAKU.2017.890917/data/index.html
エネルギー摂取量は、摂食量で調節される。
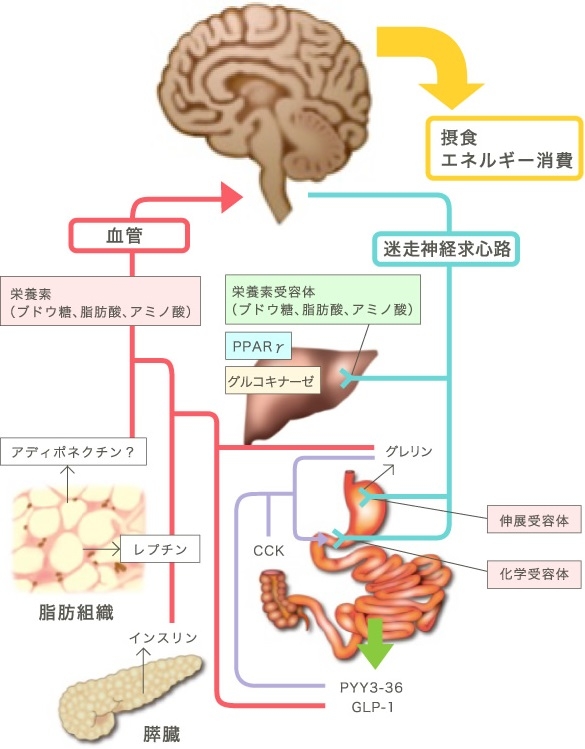
脳へのエネルギー代謝情報の入力経路は、血液と神経経路を介するものがある。
腹腔内蔵器には、迷走神経と内臓神経が分布している。腹腔内蔵器からエネルギー代謝方法を脳へ伝達する経路としては、迷走神経の構成線維の約75~90%が、内臓神経の約50%が求心性神経線維であり、この経路を使って脳へ情報を伝達する。
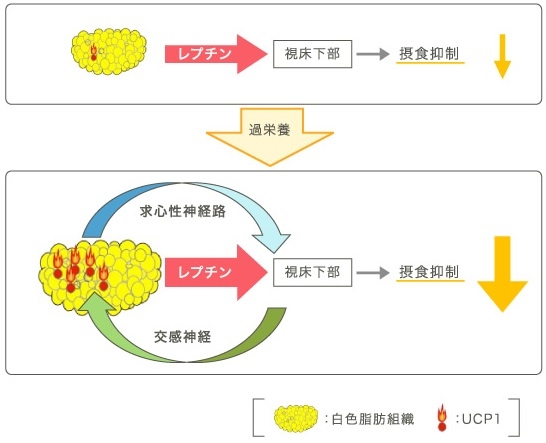
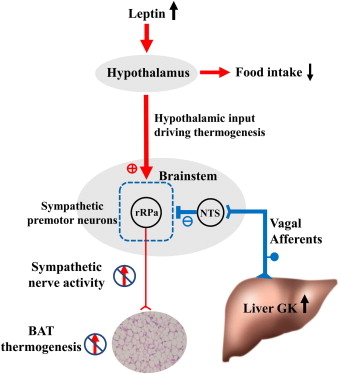
血流を介する物質としてはレプチンがある。レプチンは主に白色脂肪組織(WAT)から分泌され、血流を開始、脳の主に視床下部に作用して、摂食抑制(65%)や交感神経の活性化を介してBATの食事誘発性熱産生の増加(35%)などを引き起こす。
レプチンの産生はWATの中性脂肪の蓄積増加に相関して増える。エネルギーの過少摂取に対して体重増加を抑制するネガティブフィードバック機構として作用している。
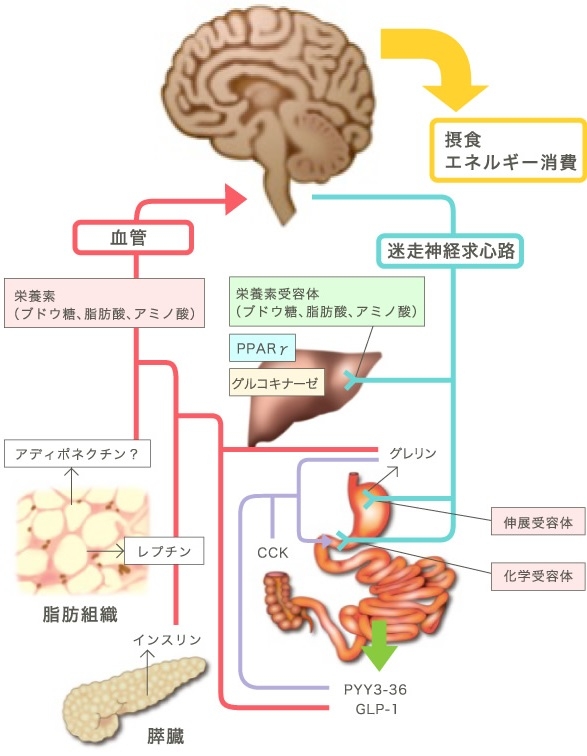
Pharmacol Ther 2008;117:188-98
レプチンが使用下部に作用し、最終的にBATに至る交感神経活性の上昇につながる神経回路は視床下部の弓状核、室傍核、背内側核などを通じて伝達され、少なくともプレモーターニューロンである延髄の縫線核に集約される。
余剰摂取エネルギーは、まず中性脂肪としてWATに著増されるが、脂肪肝は余剰エネルギーが肝細胞にまで蓄積しなければならなくなった状態=長期間のエネルギー過剰摂取を反映している。
PPARγは非肥満状態では主に脂肪組織に発現する転写因子であるが、脂肪肝では肝臓においても発現が亢進している。肝臓特異的にPPARγを欠損させると脂肪肝の形成が抑制され、末梢脂肪の増加とインスリン抵抗性や耐糖能悪化を引き起こす。
肝でのエネルギー過剰蓄積は、迷走神経求心路を介して脳に伝達され、交感神経を活性化し、BATの食事誘発性熱産生を増加させるというネガティブフィードバックにより、肥満や糖尿病の増悪を抑制している。(Science 2006;312:1656-9)
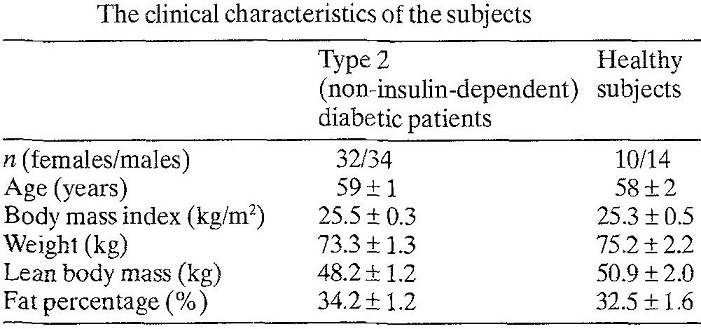
66人の二型糖尿病患者と年齢、体重をマッチングさせた健康対象者24人に対いて比較検討した臨床研究がある。二型糖尿病患者の方が基礎代謝は102.8±1.9 vs 90.7±2.8と健常者よりも高かった。8人の2型糖尿病患者においてインスリンで12ヶ月治療をすると明らかに基礎代謝は低下した。肝臓における糖新生は2型糖尿病で1044.0±29.9 vs 789.3±41.7と健常者より高かったが、インスリン治療により糖新生も低下した。
体重もインスリン治療により75.6±4.9 vs 79.5±4.9㎏と明らかに増加した。インスリンはエネルギー貯蓄に作用するホルモンといえる。
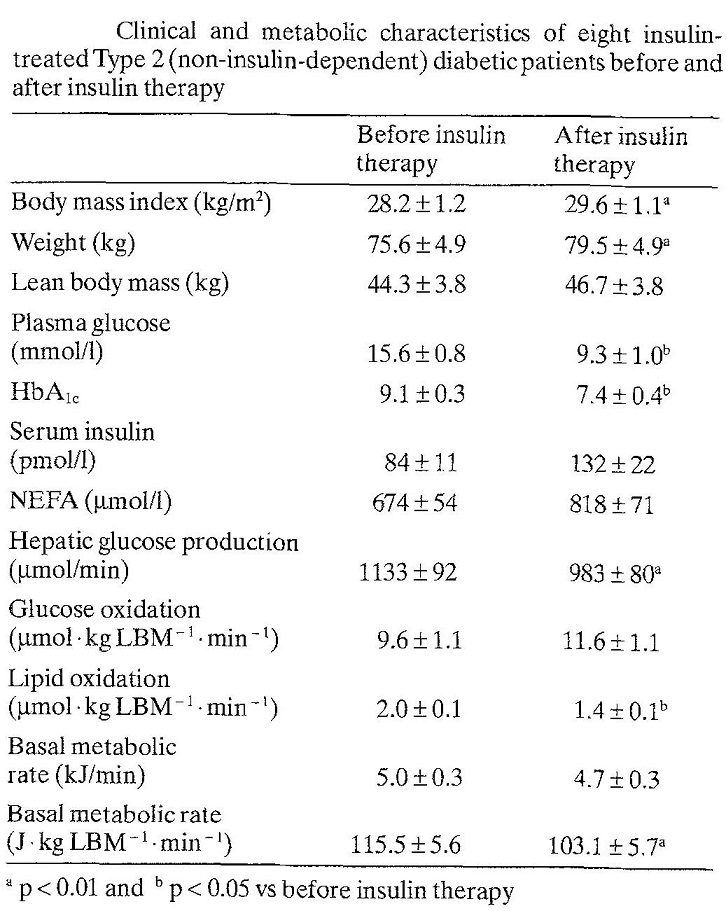
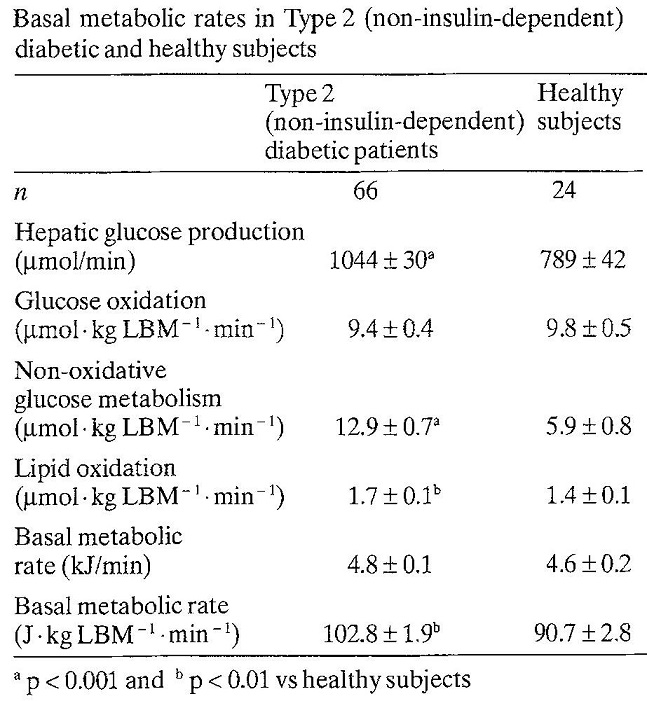
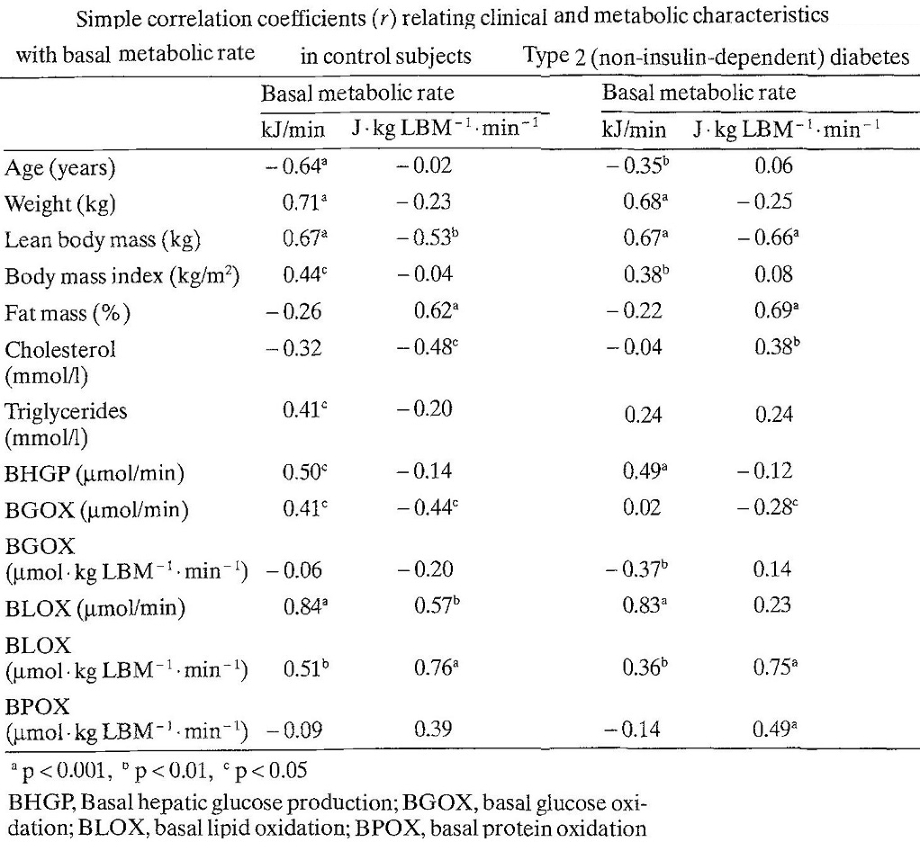
Diabetologia 1992;35:962-966
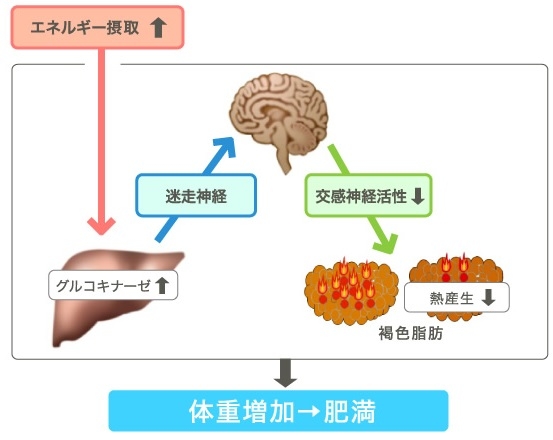
そこで我々は肝臓のグルコキナーゼ(GK)に注目した。
肝臓特異的にGKを過剰発現させたトランスジェニックマウスの血糖値はコントロールより上昇し、高脂肪食でも変化はなかったが、血中インスリン値は上昇し、その結果体重も増加した。
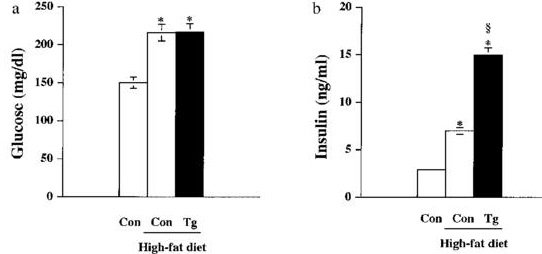
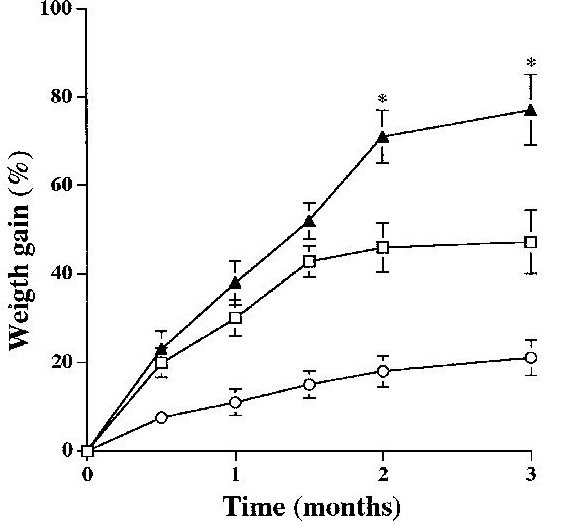
Effect of a high-fat diet in body weight gain of transgenic mice overexpressing GK in liver. Changes in body weight of control mice fed a standard diet (○), and control (□) and transgenic (▲) mice fed a high-fat diet for 3 months are shown. Data are means±SEM of 12 mice for each group. *p<0.05 Tg-fat vs. Con-fat
長期間GKの発現が亢進すると脂肪肝が出現する。
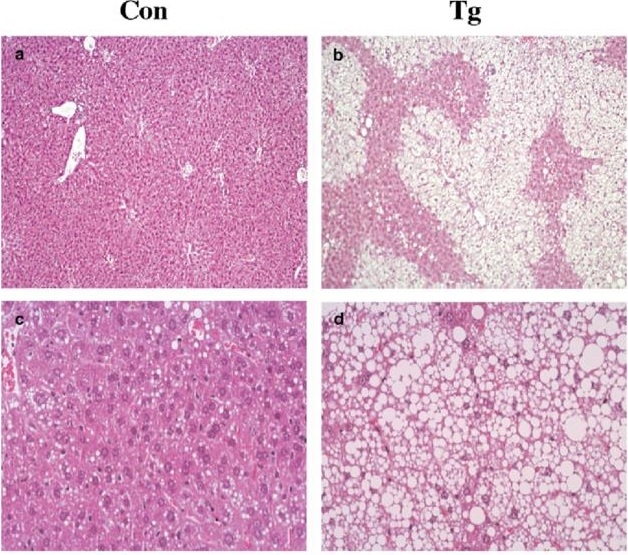
Diabetologia 46(12):1662-8
GK発現上昇は、交感神経の活性低下を経由してBATの熱産生を抑制する、ため脂肪滴が増大する。UCP1は減少し、酸素消費は1/3ほど低下し、体重が上昇してくる。
Yamada T. Inter-organ communications mediate crosstalk between glucose and energy metabolism. (Review) Diabetol Int. 4(3):149-155. 2013
GK亢進の情報は迷走神経を経由していることの検証を、肝門部の迷走神経を切断してその変化を確認した。
肝GKを特異的に発現亢進したマウスではBATでの熱産生関連遺伝子の1つであるUCP1の発現の減少がおこり、体重が増加する。高脂肪食によりさらなる体重の増加が起こる。肥満耐性マウスに肝GKを過剰発現すると体重の増加を来すが、体重増加するマウスにおいて肝GKをKnockdownすると適応性熱産生の増加とともに体重が減少する。
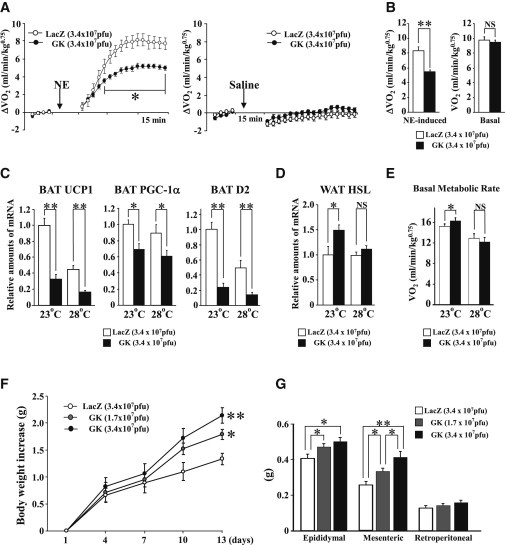
Hepatic GK Expression Suppresses BAT Thermogenesis and Increased Adiposity
Afferent Vagus Transmits Signal(s) from Hepatic Glucokinase to the Brain
Cell Metab 2012 16 825-32
人においてはinactivating glucokinase(GCK) mutationsのある患者MODY2の患者のプロファイルを見てみると、血糖値とHbA1c値は、糖尿病とControlの中間の値だが、体重は両群よりも少ない。
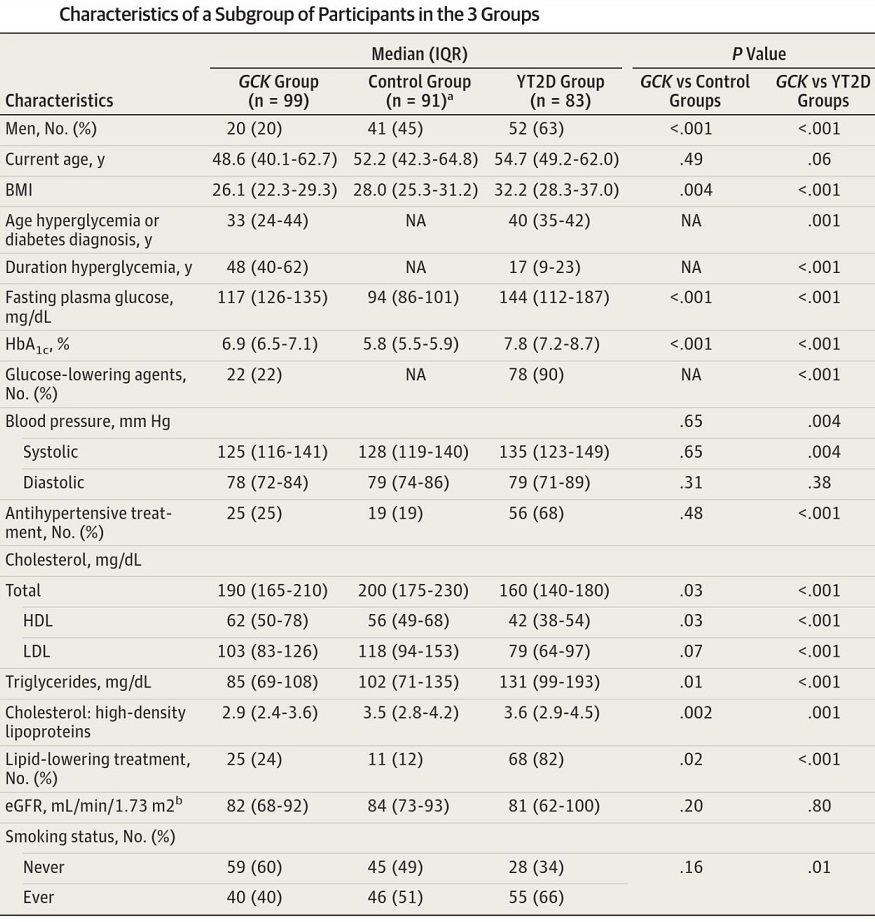
JAMA 2014;311:279-286
高インスリン血症はGKを上昇させ、体重が増加してくる。
血糖と体重をコントロールの両立を達成するためには、必要最低限のインスリンで良好な血糖コントロールする必要がある。
参:若年発症成人型糖尿病(MODY:maturity-onset diabetes of the young)
MODYは常染色体優勢で発症する若年糖尿病であり、糖代謝にかかわる単一遺伝子の障害が原因となって糖尿病を発症する疾患で、今日までに13種類が報告されている。
全糖尿病の1~3%の頻度と考えられている。欧米ではMODY2(GCK変異)とMODY3(HNF-1A変異)が主であるが、日本ではMODY3が多いと考えられていたが、最近の検討ではMODY2も多いことがわかってきた。
MODY2は血糖値に比してインスリン分泌閾値が高いことが特徴であり、空腹時血糖値の上昇は見られるが、食後血糖値や経口血糖負荷試験に時間血糖値は糖尿病域でないことも少なく、インスリン分泌能は保持されている。大半は無治療あるいは食事・運動療法で治療され予後は良好である。
https://www.shouman.jp/disease/details/07_01_003/
OGTTにてnormal glucose tolerance(NGT:耐糖能正常)の症例を上段、impaired glucose tolerance(IGT:耐糖能異常)の症例を中段、初診時HbA1c 12.3%とコントロール不良で食事療法を含めた生活習慣改善を目的として教育入院をした2型糖尿病の症例の持続血糖モニター(continuous glucose monitoring:CMG)を下段に示した。
耐糖能の悪化に伴い食後血糖の上昇と、上昇時間の長時間化が認められる。
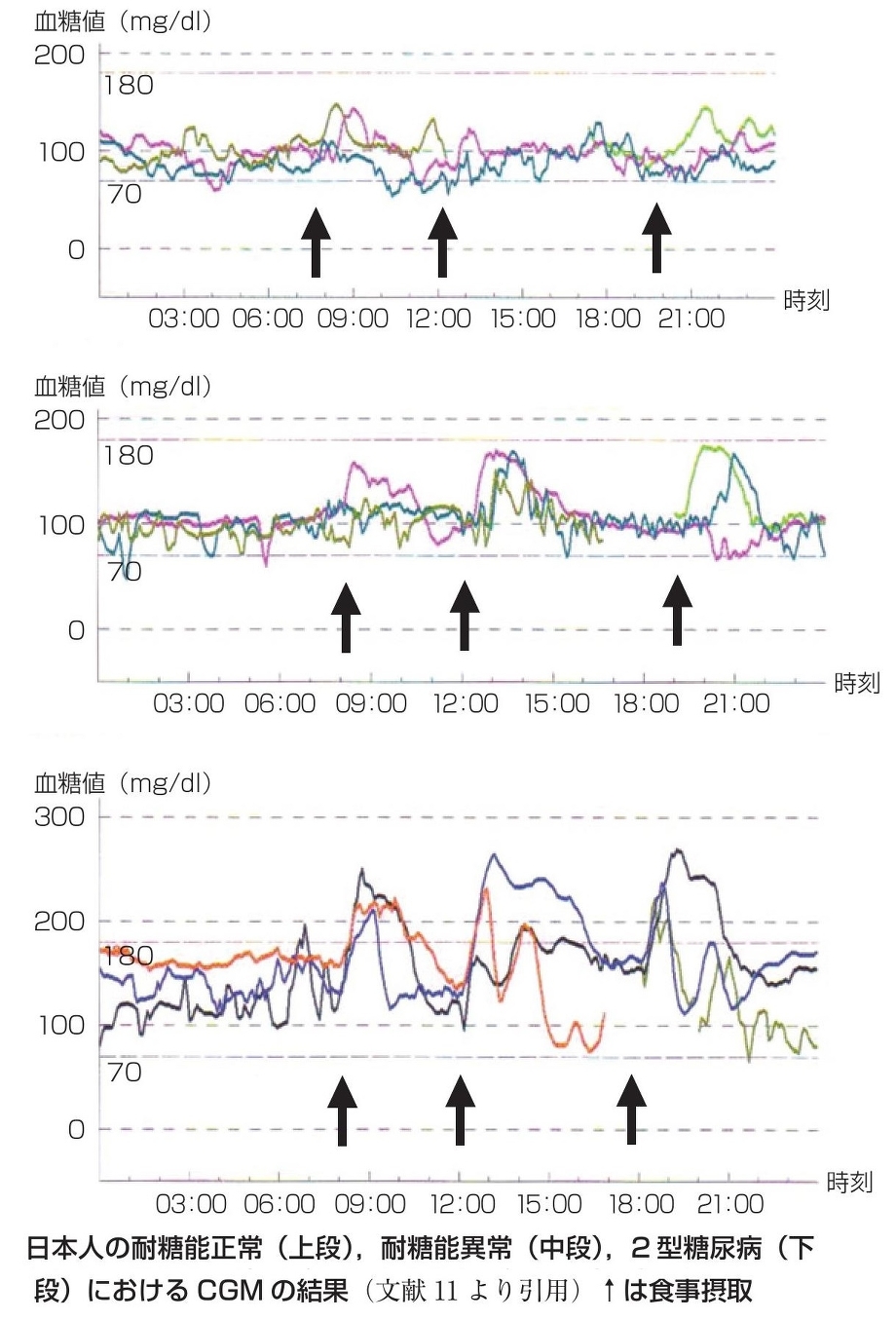
内科学会雑誌 2009;98:802-808
GLP-1作動薬であるDulaglutideを日本人2型糖尿病に投与した結果、体重が減少することが示された。
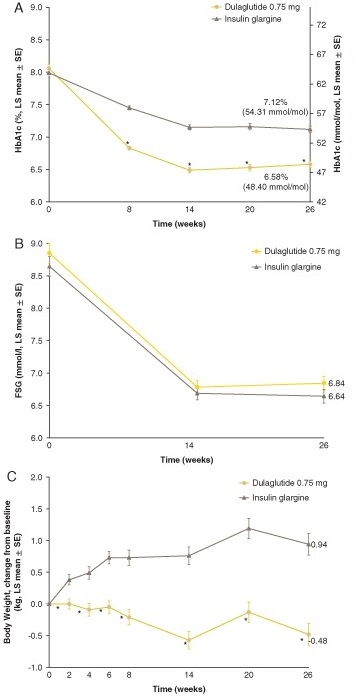
Diabetes Obes Metab. 2015 Oct; 17(10): 994-1002
糖尿病治療のガイドラインは以下のように記載されている。
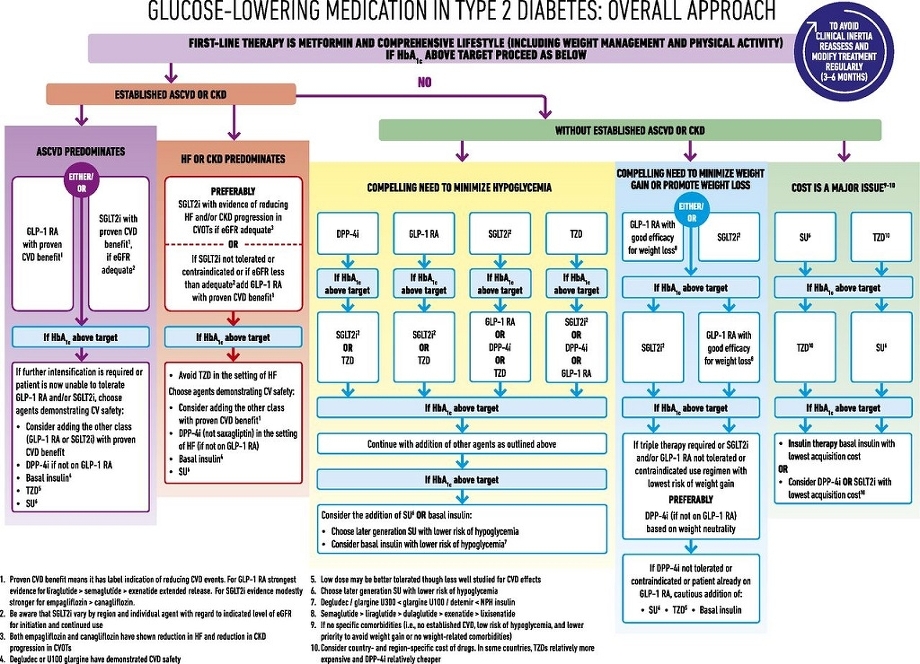
体重を増やしたくない、減らしたい患者に対しては以下のように,GLP-1製剤か、SGLT2阻害薬の投与が推奨されている。
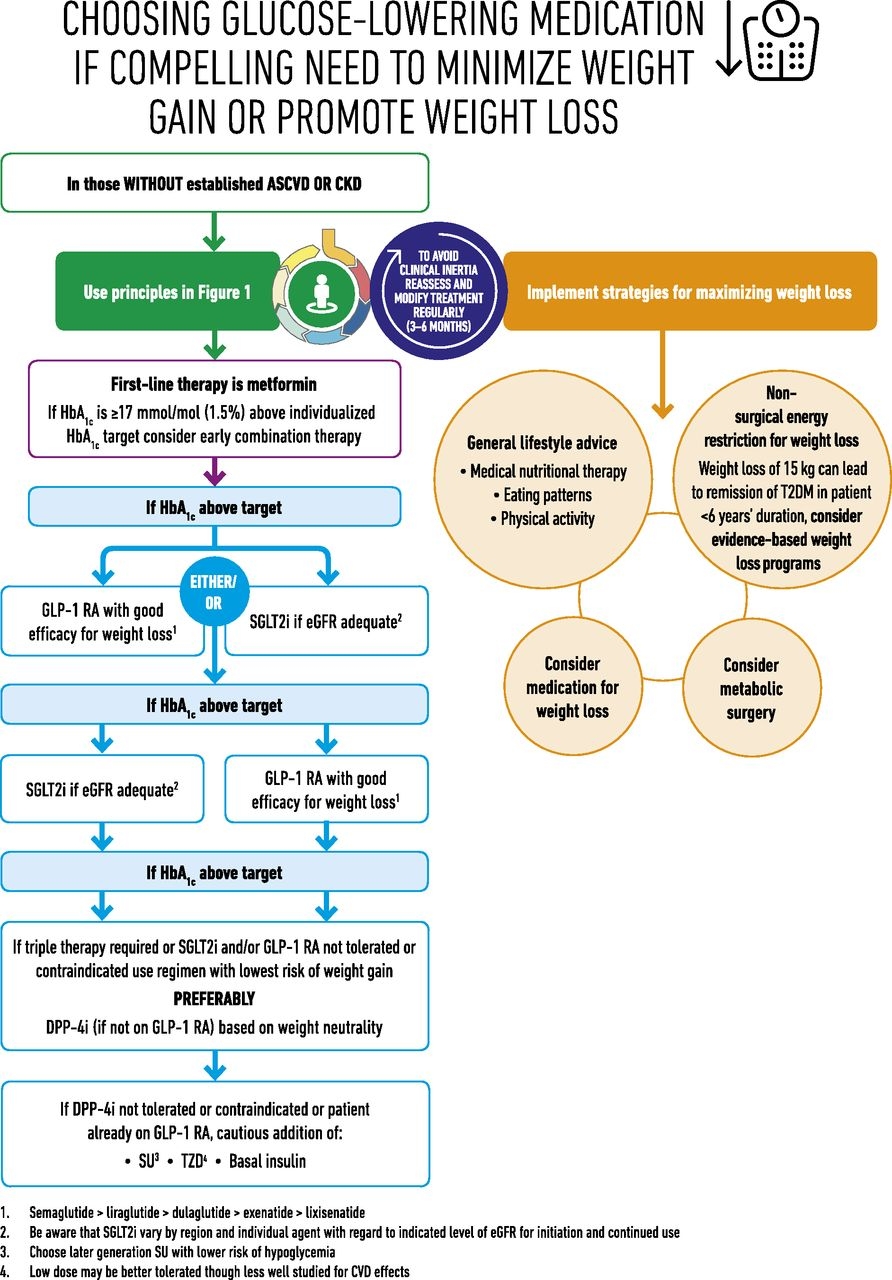
https://care.diabetesjournals.org/content/41/12/2669.supplemental
食事の摂取量を調節している物質としてレプチンがある。
Pharmacol Ther 2008;117:188-98
レプチンは1994年遺伝的に過食で肥満を発症するマウスに、正常マウスの血液を輸血すると肥満マウスの食欲が低下することから発見された肥満遺伝子obがクローニングされ、その遺伝子産物としてレプチンが発見された。(Nature 1994;372:425-432)
レプチンをob/obマウスに投与すると摂食量が減り肥満が改善する。レプチンは脂肪組織量に比例して脂肪細胞から分泌され、血流を介して主として視床下部弓状核に分布するレプチン受容体を介して摂食抑制および、エネルギー消費亢進作用を発揮する。
我々の体重は摂食量だけでなく、摂取したエネルギーをどのように利用するかによっても変化する。
例えば、強い食事制限を行うと肇はすぐに体重は減少するが、その後体重減少は緩やかとなり、元の良よりも少ない量でも、摂取カロリーを増やすと体重が逆に増加するリバウンド現象が起こる。これは、摂食量の減少に整体が適応し、身体全体のエネルギー消費が低下するためである。 摂食量が低下すると、脂肪組織量が減り、血中レプチン濃度が低下するためである。
参:レプチン抵抗性
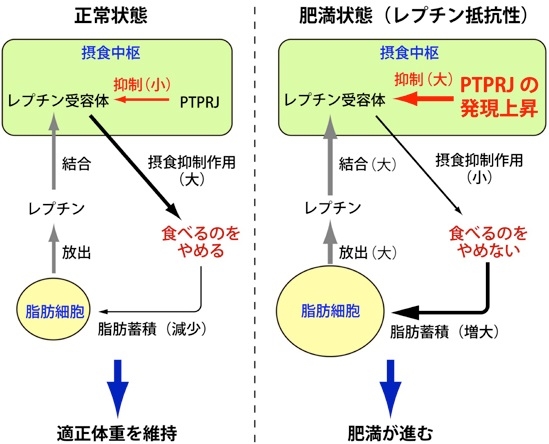
肥満状態においては、レプチンが高濃度でありながら、摂食は必ずしも抑制されておらず、レプチンが効きにくくなるレプチン抵抗性と呼ばれる現象が生じている。
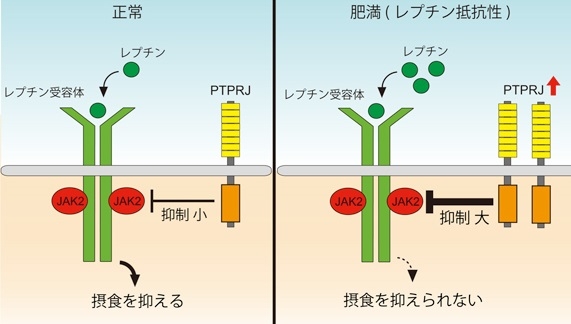
レプチン受容体にはJAK2糖蛋白質リン酸化酵素が会合している。レプチンが結合するとJAK2は自身の特定のチロシンを自己リン酸化して活性化し、さらにレプチン受容体をリン酸化することによって細胞内へレプチンの情報を伝達する。この情報伝達により、摂食中枢の神経細胞は摂食を抑制する。受容体様蛋白質チロシン脱リン酸化酵素(RPTP)の1つであるPTPRJがインスリン受容体を脱リン酸化することにより、その働きを抑制している。
PTPRJ欠損マウスでは、食後の血糖値の上昇が穏やかで、インスリンの働きが増大している。
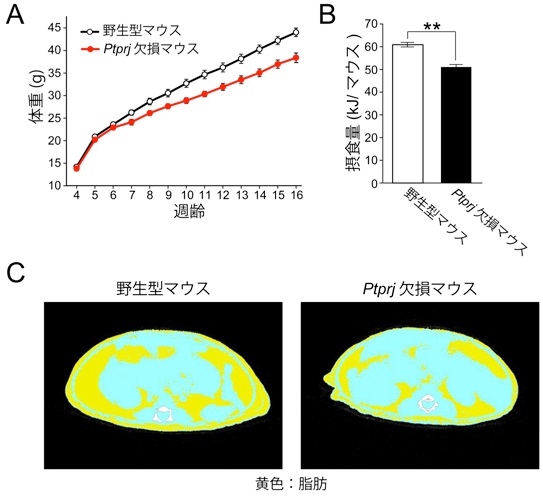
PTPRJは脳の摂食中枢の神経細胞において高発現している。Ptprjの遺伝子欠損マウスは正常に生育し、野生型マウスに比べ体調は変わらないが、摂食量が少なく、低体重である。
これらのマウスにレプチンを投与すると野生型マウスに比べて、Ptprj遺伝子欠損マウスでは、摂食量と体重が顕著に減少した。Ptprj遺伝子欠損マウスではPTPRJが無いために、レプチンの摂食抑制の働きが強まっていると考え、PTPRJがレプチン受容体に会合したJAK2を脱リン酸化することによってレプチンの働きを抑制していることを明らかにした。
マウスを高脂肪食で二か月間飼育してレプチン抵抗性を形成する。この時摂食中枢のPTPRJの発現が上昇している。
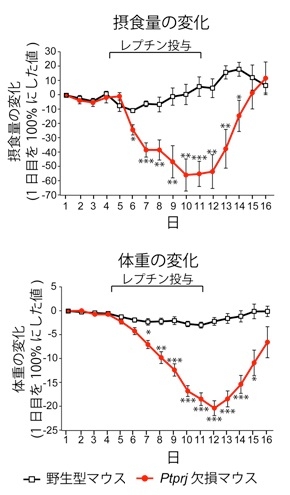
高脂肪食を長期間(14週間)投与したマウスにレプチンを投与したところ、野生型ではレプチン抵抗性を発症しており、摂食量と体重の減少がほとんど見られなかったが、Ptprj遺伝子欠損マウスではレプチン投与に応答し摂食量および体重の顕著な減少がみられ、レプチン抵抗性が生じていない。さらに、レプチン抵抗性を発症していない野生型マウスの摂食中枢に人為的にPTPRJの発現を増加させるとレプチン抵抗性が誘導された。
肥満に伴い視床下部弓状核でPTPRJの発現が症状することによりレプチン抵抗性が出現していると考えられる。
以上をまとめて図示すると以下のようになる。
http://www.nibb.ac.jp/press/2017/09/14.html
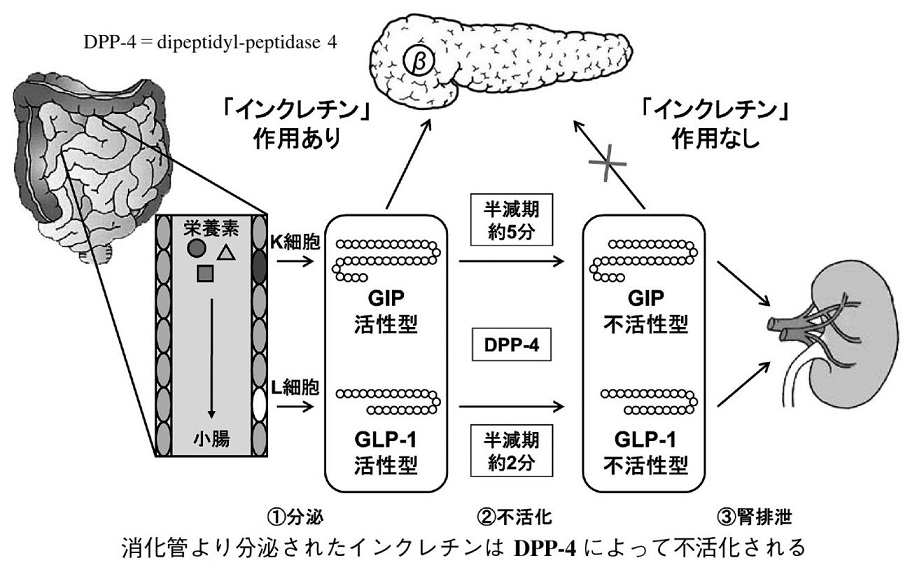
インクレチン
消化管で産生され、食事に伴って増加し、膵β細胞からのインスリン分泌を促進するホルモンの総称。
上部消化管のK細胞から分泌されるGlucose-dependent insulinotropic polypeptide(GIP)と下部消化管のL細胞から分泌されるglucagon-like peptide-1(GLP-1)の2つのホルモンがインクレチンとして機能することが確認されている。
血中のブドウ糖濃度が同じになるようにぶどう糖を経口投与した時の方が、静脈投与した時よりはるかに大きなインスリン分泌反応がみられた。
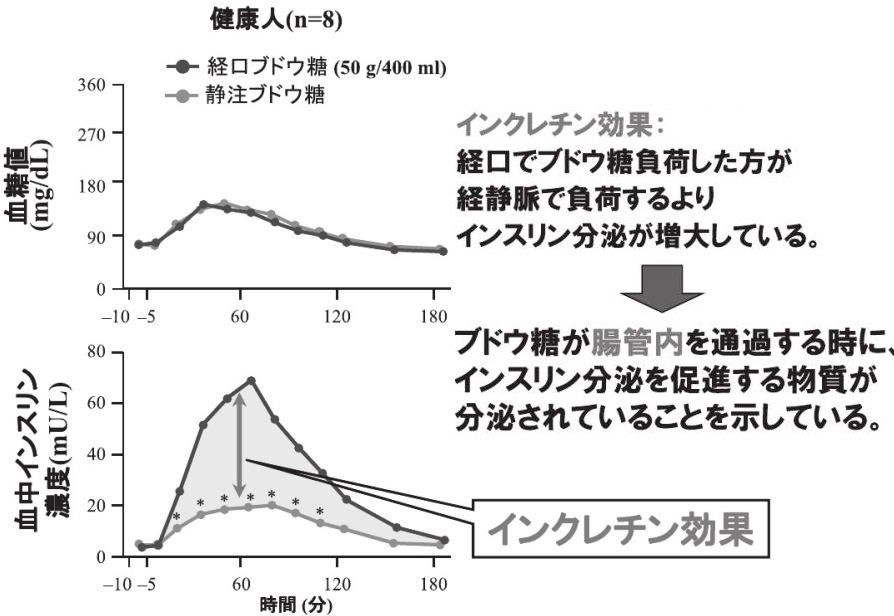
Diabetologia 1986;29:46-52
血糖値によりインスリン分泌が調節されている。腸管から吸収されたぶどう糖は門脈を経て膵β細胞に取り込まれる。膵β細胞に取り込まれたぶどう糖は解糖系ならびにミトコンドリアの電子伝達系による代謝を経てATPを産生する。ATPは細胞膜のATP感受性カリウムチャンネルの閉鎖、膜の脱分極、電位依存性カルシウムチャンネルの開口により細胞内カルシウム濃度を上昇させ、インスリン分泌が増強される。これがインスリン分泌の惹起経路である。インスリンの基礎分泌の多くが血糖値によって調節されているが、追加分泌は血糖値だけでなくインクレチンによっても調節されている。インクレチンは細胞内cAMP濃度を上昇させることでインスリン分泌を促進するが、これはインスリン分泌の増幅経路であり、惹起経路の活性を調節しているのである。
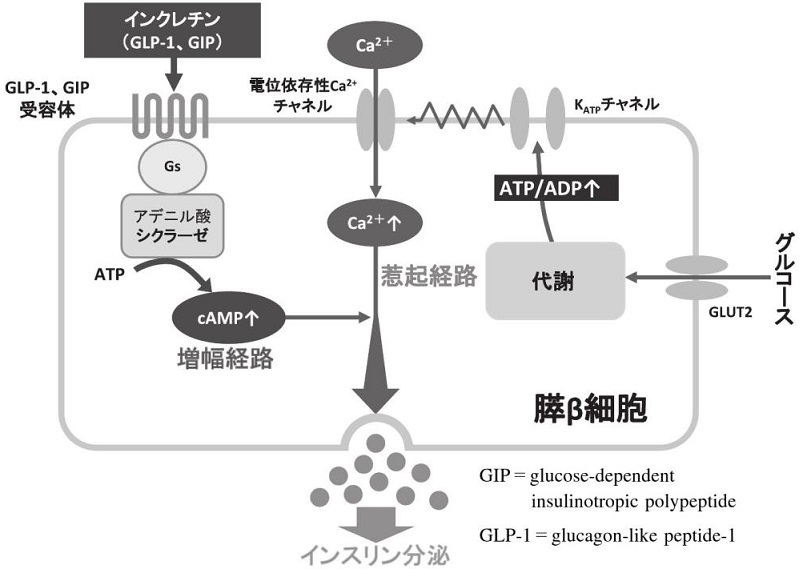
食事量が少ない時には、インクレチンの分泌は少なく、インスリン分泌は少ない。食事量が多い時は、血糖値が上昇するから、インスリン分泌が増加するのではなく、インクレチン分泌が多いために同じ血糖値でもより多くのインスリンを分泌することができるのである。
2型糖尿病患者においては、GIP分泌は健常者と変わりないが、外来性のGIPは2型糖尿病患者ではインスリン分泌促進効果は認めなかった。一方、肥満および2型糖尿病患者での食後のGLP-1分泌は障害されているが、薬理量の外来性のGLP-1によるインスリン分泌は2型糖尿病患者でも促進された。
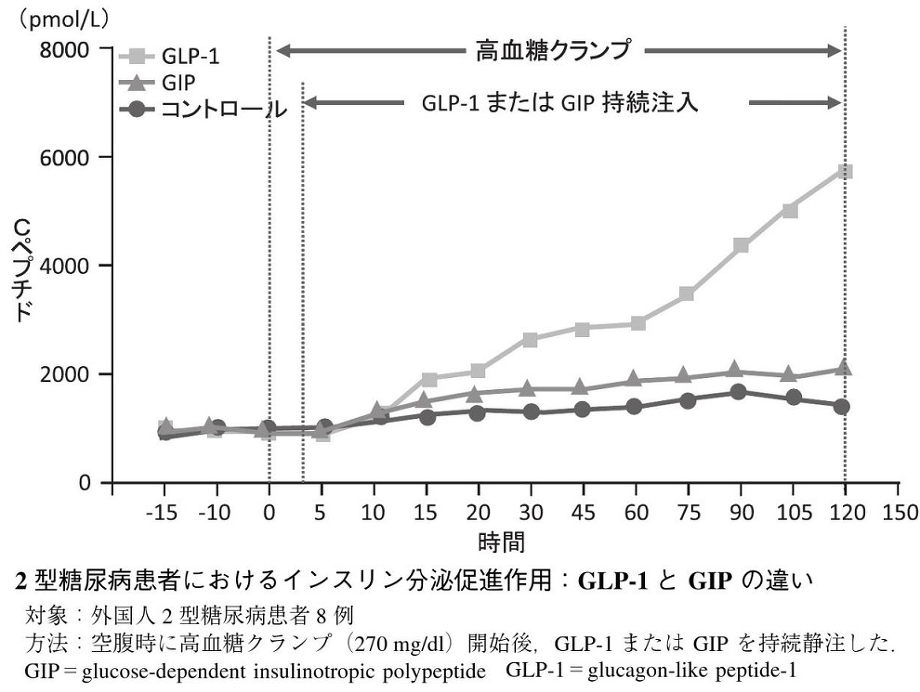
Diabetologia 2002;45:1111-1119
GLP-1およびGIPともペプチドホルモンであり、血中の酵素であるDPP-4によりただちに分解され、その半減期は2~5分である。DPP-4により分解されないペプチドGLP-1受容体作動薬(皮下注射)とDPP-4活性を抑制するDPP-Ⅳ阻害薬が開発された。GLP-1受容体作動薬は、特異的なアゴニストであり、GLP-1シグナルのみを活性化するが、DPP-4阻害薬は、GLP-1のシグナルのみならず、GIPのシグナルも活性化する。
GLP-1は、血糖依存性にインスリン分泌を促進するために低血糖が少ないことに加え、グルカゴン分泌を抑制することから血糖正常化作用が期待されること、体重を減少させるか、少なくとも増やさないことが報告されていること、血糖降下作用以外に直接血管内皮細胞機能の改善作用や、心筋保護作用などの膵外作用が期待されている。
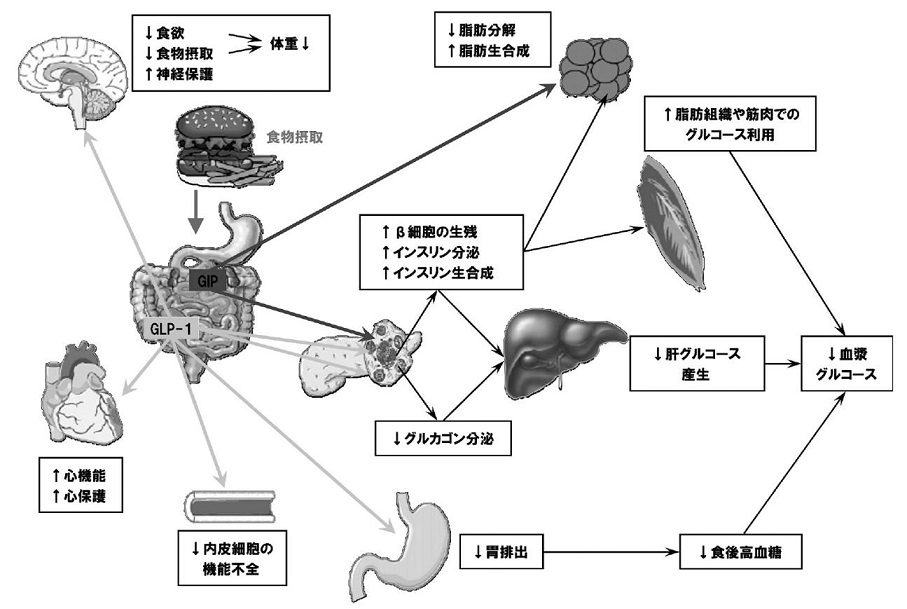
Trends Mol Med 2008;14:161-168
GLP-1作動薬とDPP-Ⅳ阻害薬の違いをまとめると以下の表のようになる。
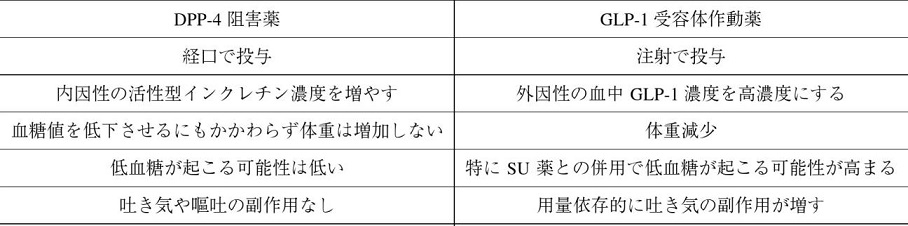
Current Medical Research and Opinion 26(5):1013-22
SGLT2阻害薬
ナトリウム/グルコース共輸送体(sodium/glucose cotransporter:SGLT)は細胞内外のナトリウムイオンの濃度差を駆動力として糖を細胞内へと取り込む能動輸送を行う。
腎層の尿細管曲部(S1セグメント)には低親和性でグルコース輸送能が大きいSGLT2が、尿細管直部(S3セグメント)には高親和性でグルコース輸送能が小さいSGLT1が存在し、腎臓の糸球体でろ過されたグルコースを再吸収している。
腎臓におけるグルコースの糸球体濾過量は一日当たり180gにも達するが、正常人ではそのほとんどが近位尿細管で再吸収される。血糖値の上昇とともに糸球体でろ過されるグルコース量も増加してくるが、近位尿細管におけるグルコースの再吸収量には限界があり、糖再吸収極量といわれ、健常男性では薬375mg/分であり、糸球体でのグルコース濾過量がこの値を超えると尿糖が出現する。
しかし、尿糖が出現する血糖値の排泄閾値は、個々のネフロンにおける再吸収量のばらつきなどの影響により、糖再吸収極量に達する前に尿糖が出現し、およそ180㎎/dL程度と考えられている。
2型糖尿病患者では、正常者と比較すると近位尿細管でSGLT2が高発現し、糖再吸収極量が上昇していることから、SGLT2の機能亢進が高血糖の維持に関与すると考えられている。
このSGLT2発現亢進がナトリウムの再吸収も亢進させている。そのため、マクラデンサへのNaClの輸送が減少し、輸入細動脈が拡張することで糸球体内圧が亢進していると考えられる。
SGLT2阻害薬を投与すると、近位尿細管での糖およびナトリウムの再吸収が抑制=ナトリウムの排泄が促進され、マクラデンサへのNaCl輸送が増加し、輸入細動脈が収縮し、糸球体内圧が低下(糸球体過剰濾過が抑制)することにより、腎保護効果がみられると考えられている。
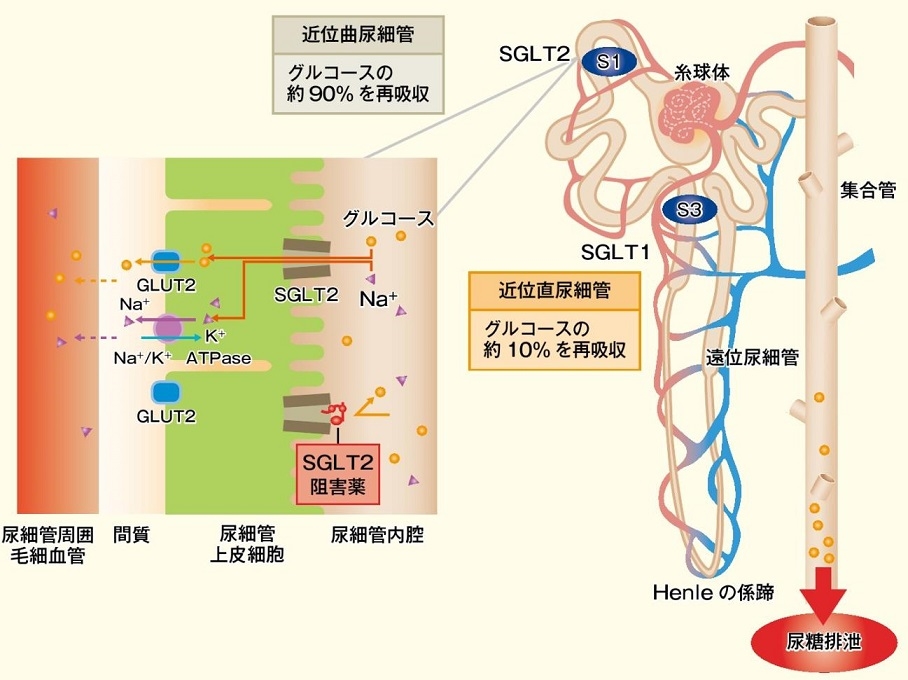
http://igaku.co.jp/pdf/1507_tonyobyo-03.pdf
糖尿病初期においては、糸球体でGFRが増加しており、近位尿細管においてSGLT2の発現が増加し、Na+/glucoseの再吸収は増加し、遠位尿細管へ移行するNa+が減少し、遠位尿細管の緻密斑(macula densa)が「Naclが減少し、GFRが低下している」と判断して、輸入細動脈を拡張させる。
SGLT2をブロックすることにより、ヘンレループ以降へのNa+/glucose輸送が増加し、緻密斑では、「NaClが増加し、GFRが増加した」と判断し、輸入細動脈拡張を解除し、糸球体内圧の低下を来して、腎保護に作用すると考えられている(tubular hypothesis)。
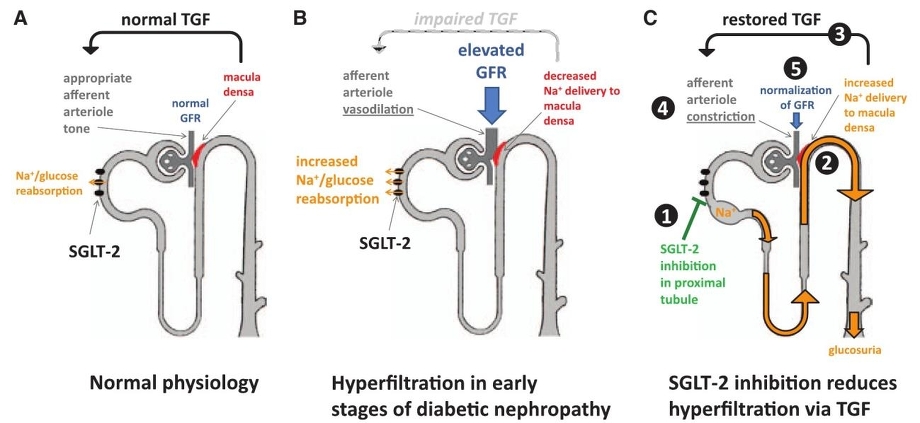
TGF(tubuloglomerular feedback mechanism):GFRが低下すると輸入細動脈の抵抗を変化させGFRを維持しようとする機構
https://www.ahajournals.org/doi/pdf/10.1161/CIRCULATIONAHA.113.005081
SGLT2阻害薬を投与して経過を見た際に、薬理学的効果から期待される体重の変化に比べ、実測した体重変化は30週を過ぎたことから頭打ちになってくる。
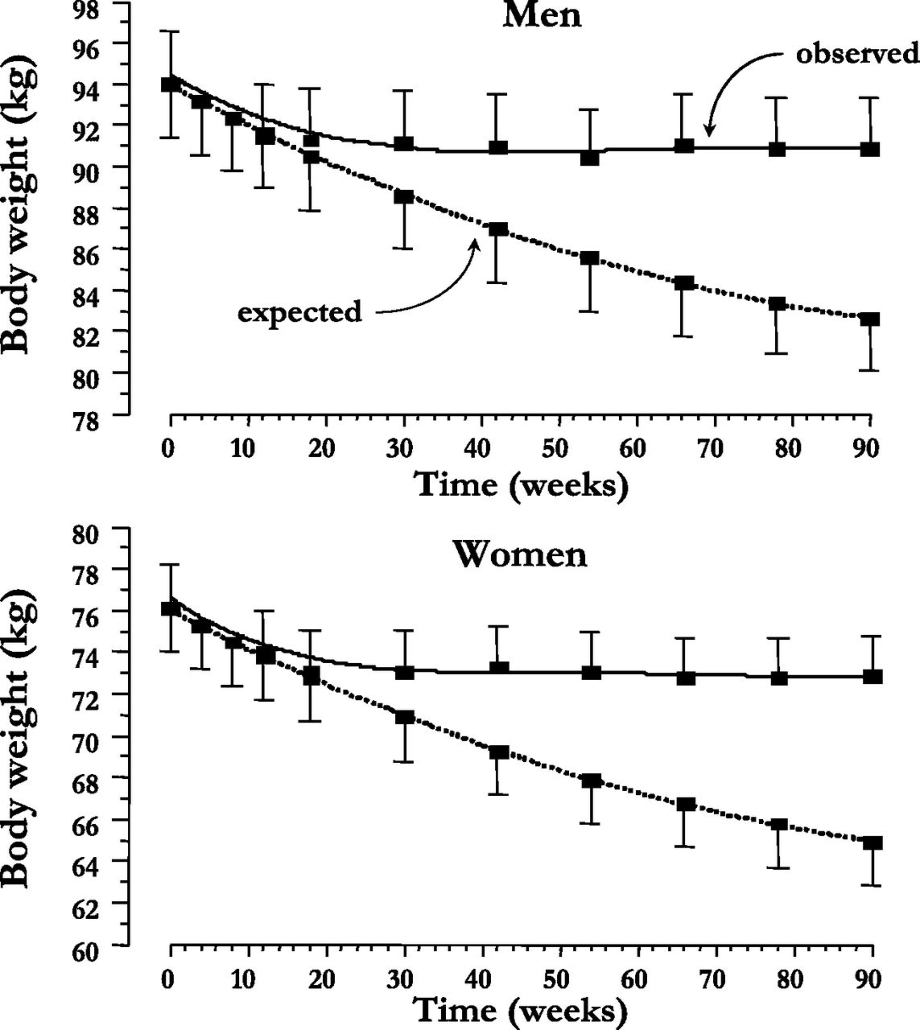
血糖値や尿中に排泄される糖の量には変化が無いが、男性においては摂取カロリーが15週を超えたころから、女性においては24週を超えたころから顕著に増加するためと考えられる。
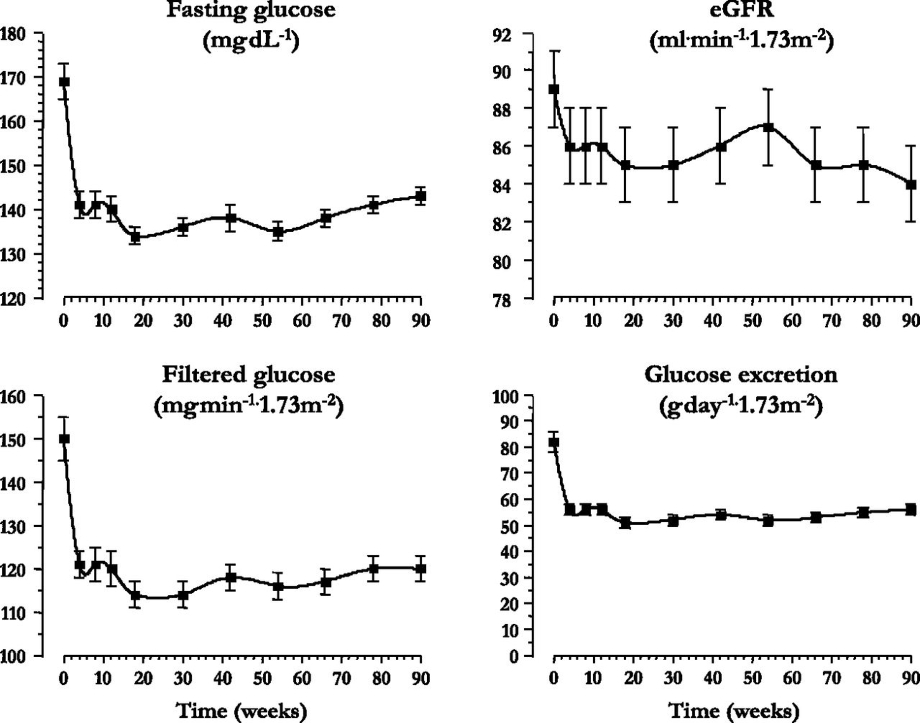
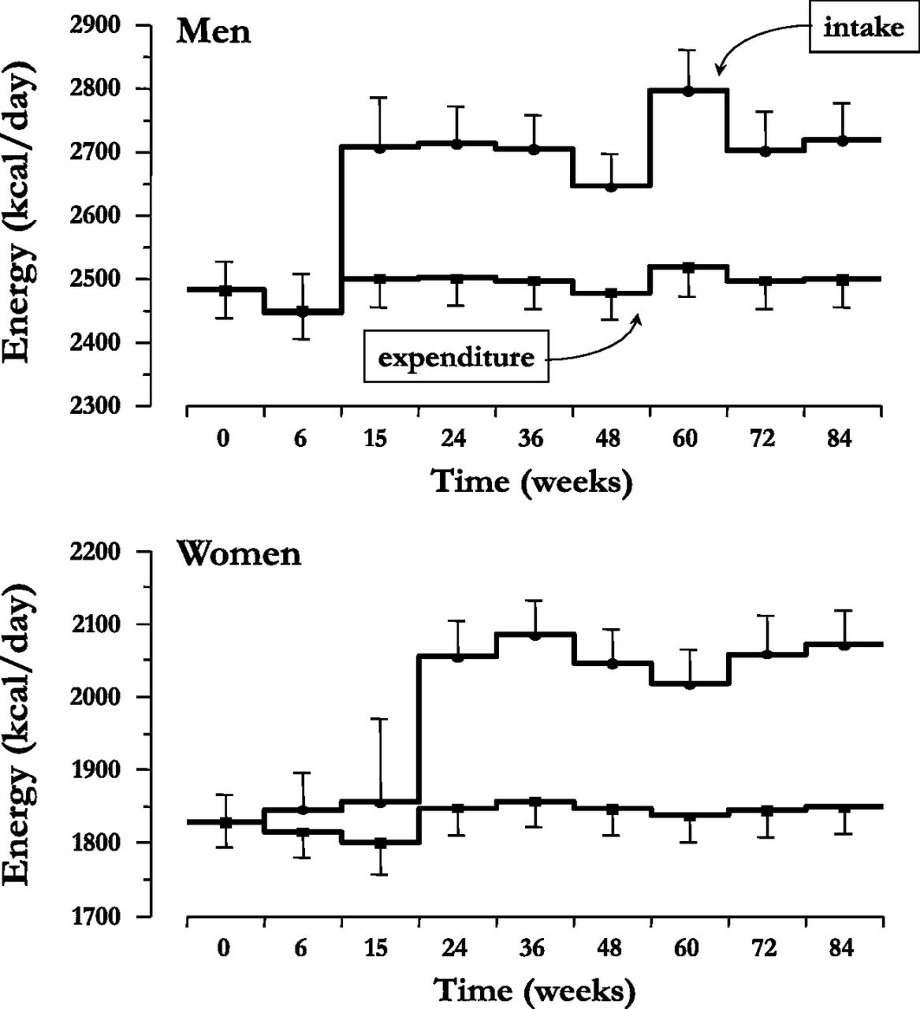
Diabetes Care 2015 Sep; 38(9): 1730-1735.
食事誘導性肥満ラットにSGLT2阻害薬投与を行い、摂食量をコントロール群と同僚として観察を行った実験では、制限摂餌した群では、自由食摂餌の群に比べ、4倍もの体重減少効果を認めた。
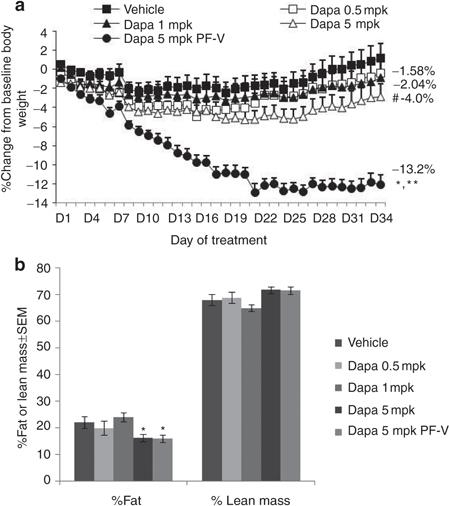
しかし、この実験でもSGLT2阻害薬投与後の約20日以降は、同程度の尿糖排泄が持続しているにも関わらず、更なる体重の減少は観察されなかった。過食の可能性以外に、SGLT2阻害薬投与が個体レベルのエネルギー消費抑制を引き起こしている可能性が考えられる。
Obesity (Silver Spring) 2012; 20: 1645-1652
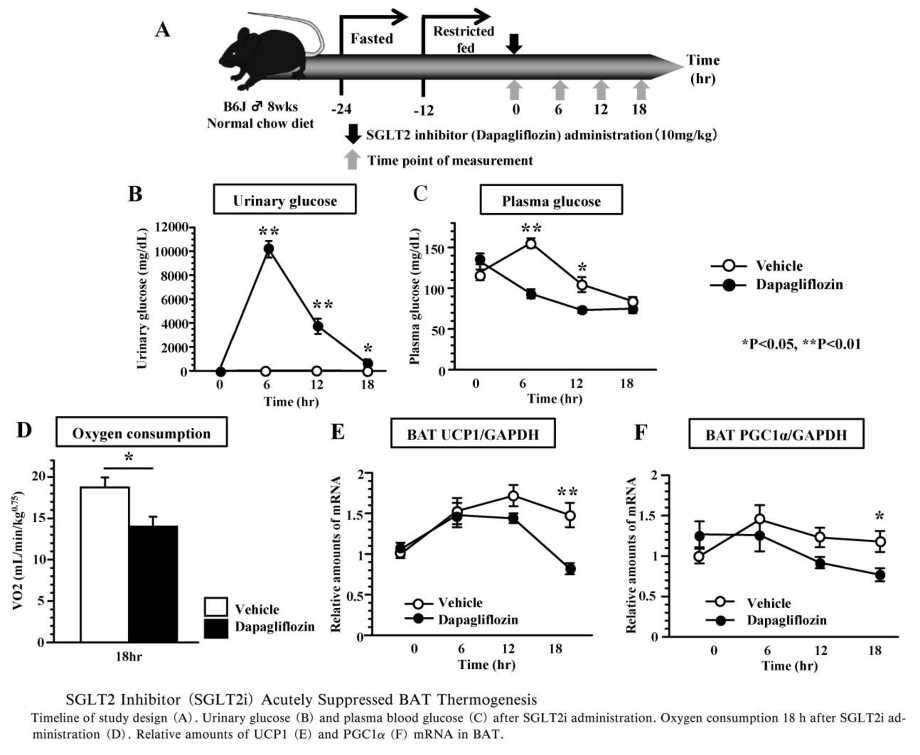
SGLT2阻害薬投与が個体の消費エネルギーに同様な影響を及ぼすかを検討した研究では、投与後18~19時間後の酸素消費量の偏倚んちはコントロール群に比べ、SGLT2阻害薬投与群で低下していた。
SGLT2阻害薬投与後のBATのUCP1発現量を測定したところ、投与後18時間後のUCP1の発現がコントロール群に比べ、SGLT2阻害薬投与群で有意に低下していた。またUCP1の発現誘導遺伝子であるperoxisome proliferators-activated receptor γ coactivator-1α(PGC1-α)の発現量も投与18時間後のSGLT2阻害薬投与群で有意に低下していた。これらの遺伝子発現はBATを支配する交感神経の活性により主に制御されているので、BATにおけるUCP1やPGC1-αの発現低下は、SGLT2阻害薬投与により、BATを支配する交感神経活性が低下したことによるものと考えられる。
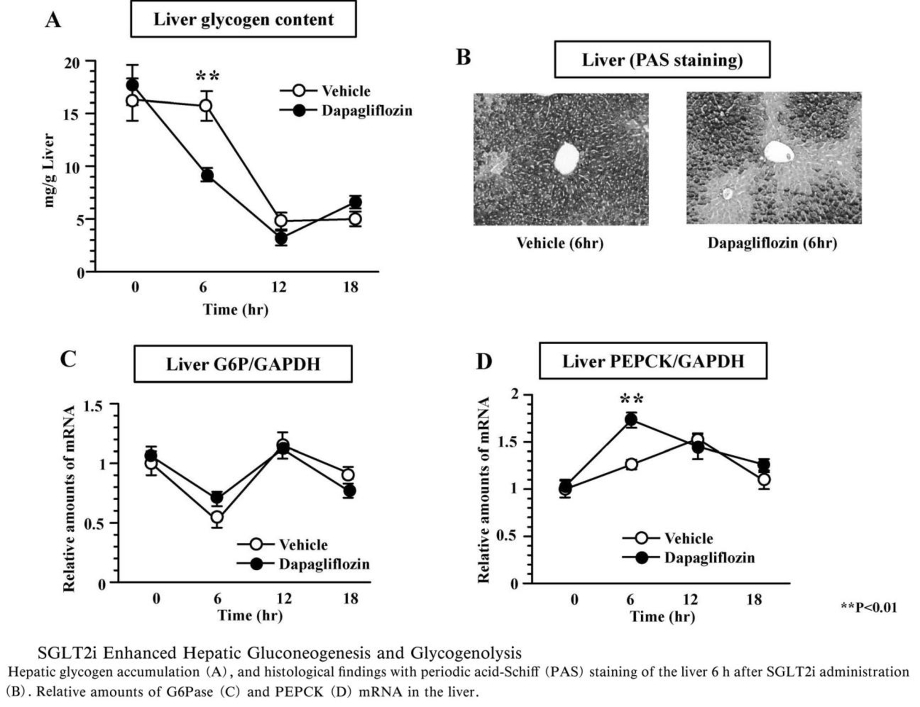
SGLT2阻害薬投与後の肝臓グリコーゲン含量測定では、6時間後に低下したが、12時間以降ではコントロール群との間に差はなかった。肝糖新生律速酵素であるphosphoenolpyruvate carboxykinase(PEPCK)の発現量は、SGLT2阻害薬群で6時間後にピークとなり、コントロール群より有意に上昇しており、SGLT2阻害薬投与6時間後に肝臓におけるグリコーゲン分解と糖新生が亢進していることがわかる。
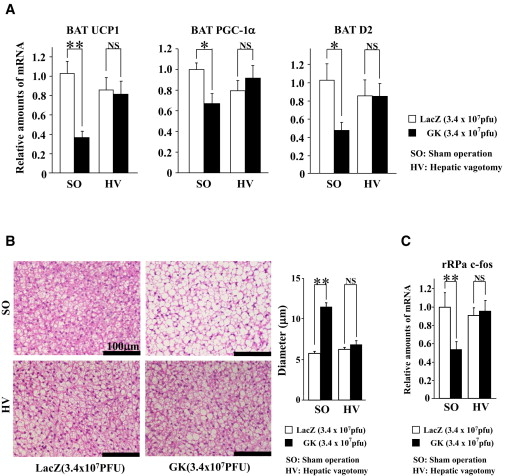
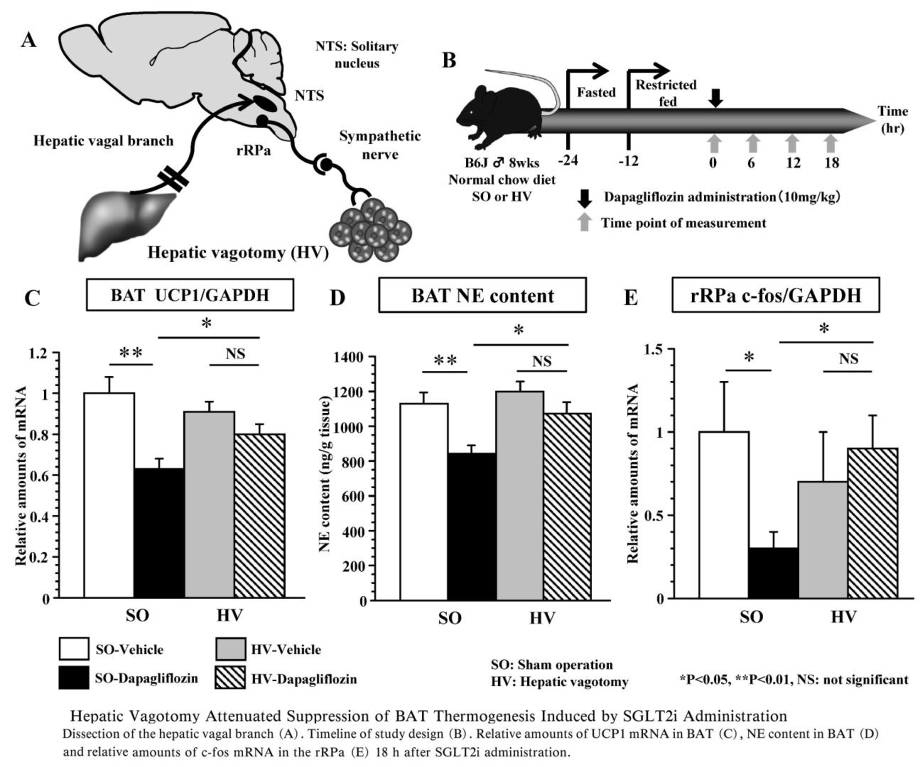
肝臓からの中枢神経への求心性神経線維を含む迷走神経肝臓枝の選択的切断を行い(selective hepatic bagotomy:HV)、絶食、制限給餌を行った後に、SGLT2阻害薬を投与しBAT熱産生に及ぼす影響をコントロール群(sham operation:SO)と比較検討した。
SO-SGLT2群でSO-コントロール群に比べ有意に認められたBATのUCP1発現量、BATのNE含量、rRPaのc-fosの発現量のそれぞれの低下が、HV-SGLT2群ではいずれも認められなかった。
以上の結果より、SGLT2阻害薬投与により肝臓から発せられるエネルギー代謝情報が迷走神経肝臓枝の求心路を介して脳へ伝えられ、その結果rRPa神経細胞の活性低下に引き続く交感神経活性の低下によってBATの熱産生が低下し、個体としてのエネルギー消費が低下したものと考えられる。
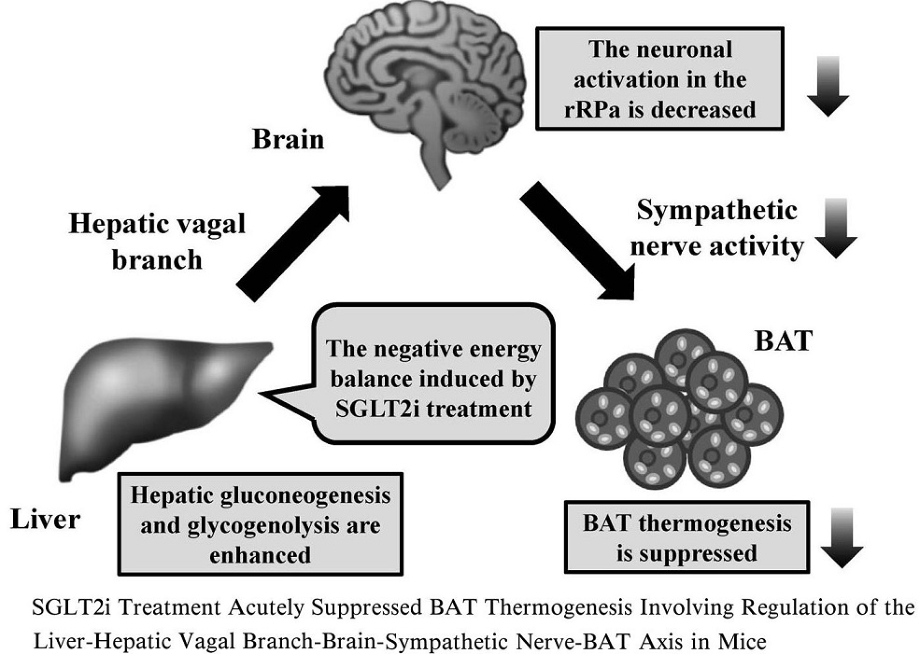
SGLT2阻害薬の投与により個体レベルでのエネルギー消費の低下が生じるのは、肝臓迷走神経枝からの求心情報が脳に伝達されrRPaの活性化が抑制され交感神経の活性低下の結果BATの熱産生関連遺伝子であるUCP1やPGC1-αの発現が抑制され、BATの熱産生が抑制されると考えられる。
カテゴリ一覧
最近のブログ記事
月別アーカイブ

「生活習慣を改善して健康な身体になりたい」
「病気ではないけれど健康に不安がある」




















